
When a factory produces a batch of medical devices and 12 out of 500 units fail inspection, what happens next? Stopping the line and tossing the bad parts isn’t enough. That’s a correction. What really matters is figuring out why those 12 failed in the first place-and making sure they never happen again. That’s corrective action, and it’s the backbone of quality in modern manufacturing.
What’s the Difference Between a Correction and a Corrective Action?
A correction is like putting a bandage on a cut. You fix the immediate problem: you scrap the defective parts, rework the assembly, or adjust a machine setting. Quick. Simple. But it doesn’t stop the bleeding. Corrective action is surgery. It cuts deep. It asks: Why did the machine drift out of tolerance? Why did the operator skip the calibration check? Why did the supplier send material with inconsistent hardness? The goal isn’t just to fix today’s problem-it’s to kill the cause so the problem can’t come back. In regulated industries like medical devices and pharmaceuticals, regulators don’t just want you to fix things. They want proof you understand why they broke. The FDA’s 2023 warning letters show that 61% of companies failed because they treated symptoms instead of root causes. That’s not negligence-it’s a misunderstanding of the system.The Six-Step CAPA Process That Works
Every effective corrective action follows the same six-step path, whether you’re making pacemakers or car engines.- Identify the problem-It starts with data. A failed test. A customer complaint. A machine alert. At this stage, you don’t guess. You collect: serial numbers, timestamps, operator logs, inspection readings. The more detail, the better.
- Evaluate the risk-Not all defects are equal. A mislabeled pill bottle? High risk. A slightly off-color paint job on a non-critical housing? Low risk. Regulators like ISO 13485 require you to categorize issues by impact on safety, performance, or compliance. This tells you how hard to dig.
- Find the root cause-This is where most teams fail. They stop at “operator error” or “bad material.” That’s lazy. Use tools like the 5 Whys: Why did the part break? Because the torque was too high. Why? Because the tool wasn’t calibrated. Why? Because the schedule didn’t include it. Why? Because no one owned the maintenance log. Why? Because there’s no accountability system. Now you’ve got something real to fix.
- Plan the fix-Your plan needs four things: specific actions (e.g., “Install torque sensor with auto-lockout”), deadlines (e.g., “Implement by Jan 15”), owners (e.g., “John Chen, Maintenance Lead”), and how you’ll verify it worked (e.g., “Run 50 units under load, track failure rate”).
- Implement and document-Don’t just change the process. Train the team. Update the work instructions. Log the change in your quality management system. Paperwork isn’t bureaucracy-it’s your defense in an audit.
- Verify effectiveness-This is non-negotiable. You can’t say “it’s fixed.” You have to prove it. Test 30+ units. Run statistical process control charts for three production cycles. If defect rates don’t drop by at least 50%, you haven’t solved it yet.
Why 68% of Companies Get It Wrong
Most quality teams think they’re doing corrective actions. They’re not. They’re doing corrections with extra steps. The biggest mistake? Skipping verification. A 2023 Cognidox study of 157 medical device firms found that 38% of CAPAs had no measurable outcome. They closed the ticket because the manager said “it looked better.” That’s not compliance. That’s wishful thinking. Another common trap: blaming people. “The operator didn’t follow procedure.” But why didn’t they? Was the procedure unclear? Was it too long? Was the training outdated? The system failed before the person did. And then there’s paperwork overload. One Reddit user, QualityEngineer2020, said their team spends 15% of their time just filling out CAPA forms. A single issue can generate 47 pages of documentation. That’s not control-it’s paralysis.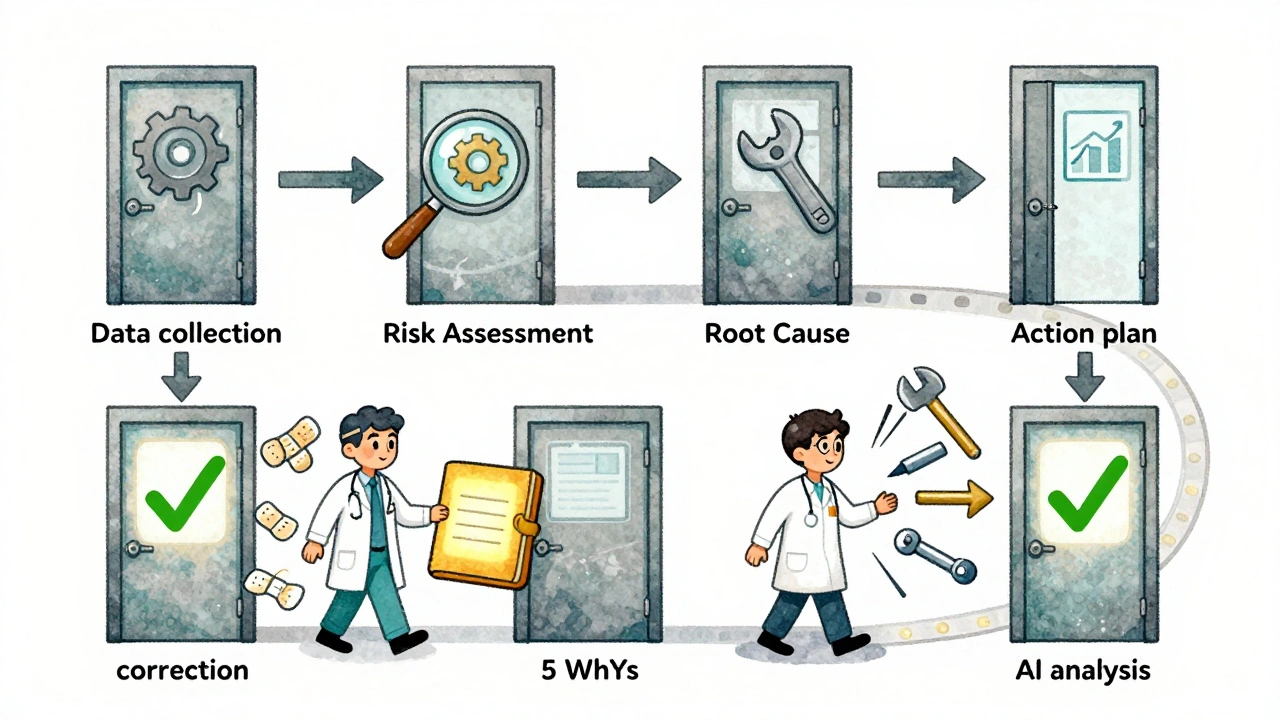
How Digital Tools Are Changing the Game
The best manufacturers aren’t using Excel sheets anymore. They’re using digital CAPA systems integrated with their production line. Tulip’s 2023 case studies show companies using AI-powered root cause analysis cut investigation time by 52%. How? The system pulls data from sensors, compares it to historical failures, and suggests the top three likely causes. Instead of eight hours of meetings, you get a report in 20 minutes. These systems also auto-generate verification reports. When you implement a new torque setting, the system automatically pulls the next 50 production runs and graphs the torque values. If they’re stable? The CAPA closes itself. No manual charting. No missed deadlines. FDA’s 2023 Digital Health Innovation Plan encourages blockchain-based audit trails. That means every change, every test, every signature is timestamped and unchangeable. No more “lost” documents. No more “I didn’t know” defenses.What Works in Medical Devices Doesn’t Always Work in General Manufacturing
Not every factory needs a full CAPA system. In medical device manufacturing, ISO 13485 requires CAPA for every non-conformity that affects safety or performance. That’s 82% of firms. In pharmaceuticals, cGMP rules are just as strict. But in small-batch metal fabrication or custom furniture making? Over-engineering the process kills speed and agility. The rule of thumb: If your product could harm someone if it fails, you need CAPA. If it’s a plastic handle on a kitchen tool? A simple correction log with a quick root cause note is enough. The key is proportionality. Don’t use a sledgehammer to crack a nut. But don’t ignore the nut either.
Real Results: What Happens When You Do It Right
In a Melbourne-based medical device maker, a recurring issue was leaking syringe barrels. For six months, they replaced the seals. Every batch had 2-3 leaks. Cost? $18,000 a month in scrap and rework. They ran a proper CAPA. Root cause? The seal supplier changed the compound without notifying them. The material had a 0.3% higher shrinkage rate. That tiny change caused micro-gaps under pressure. They switched suppliers. Updated the incoming inspection spec. Added a pressure test at the end of the line. Verified with 50 units over three weeks. Defects dropped to zero. Result? $216,000 saved in one year. No customer complaints. No FDA observations. And the quality team got a bonus.What to Do Next
If your factory is still fixing the same problems over and over:- Stop accepting “operator error” as a root cause. Dig deeper.
- Start tracking your CAPA closure rate. If more than 20% take longer than 30 days, your process is broken.
- Integrate your quality system with your production data. No more manual entry.
- Train your team to ask “Why?” five times-not just to blame, but to understand.
- Measure success by defect reduction, not paperwork completed.
What’s the difference between corrective action and preventive action?
Corrective action fixes something that already went wrong. Preventive action stops something from going wrong before it happens. For example, if a machine breaks down every three months, corrective action fixes the broken part. Preventive action adds scheduled maintenance to stop the breakdowns from ever occurring. Both are part of CAPA, but they serve different purposes.
How long should a corrective action take to complete?
There’s no universal deadline, but regulators expect timely action. For critical issues affecting safety, closure should happen within 30 days. For minor issues, 60-90 days is acceptable. The key isn’t speed-it’s effectiveness. A 90-day CAPA that works is better than a 10-day one that fails. Most successful manufacturers track average CAPA cycle time and aim to reduce it by 15% each year.
Do I need software to manage corrective actions?
You don’t need software, but you’ll struggle without it. Manual CAPA systems using spreadsheets or paper logs are slow, error-prone, and hard to audit. Digital systems automate reminders, link data from machines, and generate compliance reports. For any manufacturer with more than 50 employees or regulated products, software isn’t optional-it’s a necessity. The market for quality management software grew 11.3% in 2022, and for good reason.
What happens if I don’t do corrective actions?
In regulated industries, you risk FDA warning letters, product recalls, or even shutdowns. In 2022, 28% of all FDA quality system observations were for poor CAPA implementation. Outside regulated spaces, you’ll lose customers. Repeated defects mean lost trust, higher returns, and higher costs. One automotive supplier lost a $12M contract because they couldn’t prove they fixed a recurring sensor failure.
How do I know if my corrective action worked?
You measure it. Look at your defect rate before and after the fix. Use statistical tools like control charts. If the defect rate drops by at least 50% and stays low for three full production cycles, you’ve likely succeeded. If defects return, you didn’t fix the root cause. Don’t trust gut feelings. Trust data.
Joel Deang
December 3, 2025 AT 17:32Roger Leiton
December 4, 2025 AT 19:52Laura Baur
December 5, 2025 AT 07:11Steve World Shopping
December 6, 2025 AT 05:05Lynn Steiner
December 8, 2025 AT 04:31Alicia Marks
December 8, 2025 AT 18:48Paul Keller
December 10, 2025 AT 15:44Shannara Jenkins
December 12, 2025 AT 12:41Elizabeth Grace
December 14, 2025 AT 03:04Steve Enck
December 14, 2025 AT 06:44